Atrésie de l’œsophage : de l’inquiétude à la connaissance
Besoins d’information des parents et considérations sur l’étiologie, les soins et le soutien parental
Auteurs : Katherine Merritt Doucet a,b, Juliana Coronado a, Rebecca Leslie c, Emily Whitesel d,e, Benjamin Zendejas c,e, Dusica Bajic a,e,*
Traduction par Sabine Grataloup de la publication « Esophageal Atresia: From worry to wisdom–Parental information needs and considerations on etiology, care, and parental support » paru le 22 Novembre 2025 dans la revue Global Pediatrics.
AVERTISSEMENT : cette traduction n’est fournie qu’à titre indicatif, le texte de référence de cette étude est celui d’origine en anglais dont le lien est fourni ci-dessus.
Résumé
L’atrésie de l’œsophage (EA) est l’une des anomalies gastro-intestinales congénitales les plus courantes, posant des défis complexes en matière de diagnostic, de traitement et de soins à long terme. Cette revue narrative aborde quatre domaines clés : l’étiologie et le diagnostic, les techniques de réparation chirurgicale, les défis liés à l’alimentation, ainsi que le stress et le soutien des parents. Elle présente les données actuelles et les considérations cliniques pertinentes pour les familles, le personnel clinique et les chercheurs. En mettant en évidence les lacunes en matière d’information et les préoccupations à long terme, cette revue contribue à améliorer la communication, la planification des soins et l’orientation de la recherche afin d’optimiser les résultats chez les enfants atteints d’AE.
Mots clés : Soins intensifs, Processus de Foker, Chirurgie néonatale, Pédiatrie, Sédation prolongée
1. Introduction
L’atrésie de l’œsophage (AE) est l’une des anomalies gastro-intestinales congénitales les plus courantes, avec une incidence allant de 1 sur 3 000 à 1 sur 4 500 naissances vivantes.77 Ses diverses manifestations anatomiques reflètent la complexité du développement trachéo-œsophagien. La classification de Gross, qui catégorise l’AE en fonction de la relation entre l’œsophage et la trachée et de la présence d’une fistule trachéo-œsophagienne (FTE), reste le système le plus largement utilisé (Fig. 1). La prise en charge thérapeutique est influencée par la longueur de la lacune œsophagienne et la présence d’anomalies associées (Tableau 1). Grâce aux progrès réalisés dans les soins chirurgicaux et périopératoires, les taux de survie se sont considérablement améliorés, dépassant 90 % chez les patients présentant des anomalies associées et approchant 100 % dans les cas d’EA isolée28, 47.
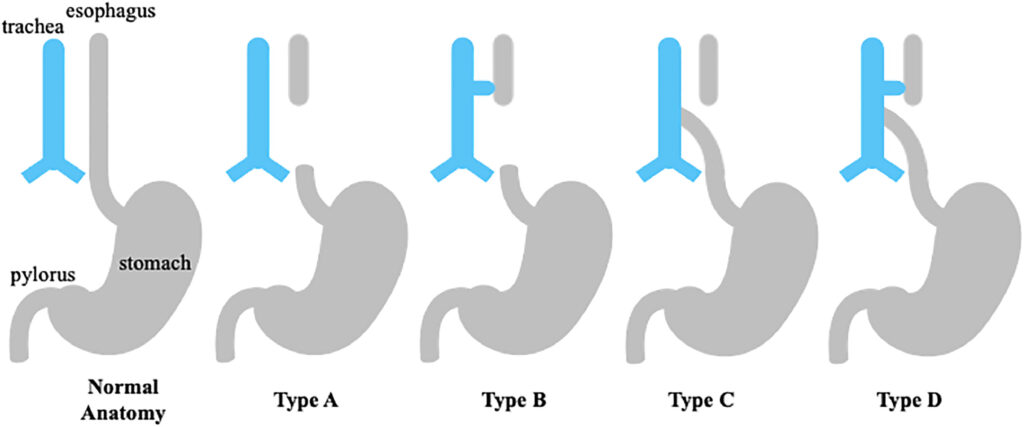
Fig. 1. Classification anatomique de l’atrésie de l’œsophage (AO) en fonction de la présence d’une fistule trachéo-œsophagienne (FTE). Le schéma illustre les sous-types anatomiques de l’AO tels que décrits à l’origine par Gross38, par rapport à l’anatomie trachéo-œsophagienne normale. Chaque sous-type met en évidence la présence et l’emplacement d’une ou plusieurs fistules trachéo-œsophagiennes et la longueur relative de la lacune atrésique. Le type C, qui se caractérise par une poche œsophagienne proximale et une FTE distale, est le sous-type le plus courant. L’AO à intervalle court, généralement de type C et D, est plus souvent associée à l’association VACTERL. En revanche, l’AO à intervalle long, observée principalement dans les types A et B, est plus susceptible de se présenter comme une anomalie isolée ou en association avec la trisomie 21 (voir tableau 1). Une cinquième variante, anciennement désignée comme type E et appelée FTE isolée sans EA, n’est plus incluse dans le système de classification standard.
De nos jours, le conseil prénatal fait partie intégrante de la planification chirurgicale et de la préparation de la famille. En effet, les parents d’enfants atteints d’AO sont souvent confrontés à des défis émotionnels et éthiques considérables. L’étude Linked European Cohort of Children with Congenital Anomalies Trial (EUROlinkCAT) souligne l’impact émotionnel subi par les familles et insiste sur l’importance d’une communication opportune et compatissante20. Des explications claires, un soutien empathique et une éducation fiable sont essentiels pour réduire l’anxiété et autonomiser les familles68. Cette revue aborde les questions clés soulevées par les parents et les soignants, en se concentrant sur l’étiologie, le diagnostic, les approches chirurgicales et les soins postopératoires, ainsi que sur le stress parental et le soutien. Pour des discussions plus détaillées sur l’AO, nous recommandons également plusieurs revues récentes et complètes71, 105.
2. Approche
Cette revue fait partie d’une étude clinique approuvée par le comité d’éthique institutionnel. Les questions des parents ont été synthétisées en six domaines thématiques (Fig. 2 ; traduction en espagnol fournie en annexe), dont quatre sont abordés dans le présent article. La liste des questions reflète notre expérience clinique collective plutôt que des données d’enquête formelles. Bien que les voix des parents aient dépassé le cadre de cette revue, les études futures devraient intégrer des méthodes qualitatives, telles que les expériences rapportées par les parents, afin de fournir un contexte plus riche et d’humaniser le parcours clinique des enfants atteints d’AO. La revue de la littérature a été réalisée à l’aide de recherches ciblées dans PubMed à l’aide de mots-clés correspondant aux préoccupations courantes des parents. Chaque section est présentée en deux parties : un aperçu général destiné à un public non spécialisé et une synthèse scientifique des données actuelles. Ce double format vise à combler les lacunes tant dans la recherche que dans l’éducation familiale, améliorant ainsi la compréhension et la communication entre les parents, les cliniciens et les chercheurs.
Fig. 2. Document récapitulatif destiné aux parents de nourrissons atteints d’atrésie de l’œsophage (AO).

Tableau 1. Caractérisation de l’atrésie de l’œsophage en fonction de la longueur de l’intervalle.
| Intervalle oesophagien* | AO à intervalle court <3 cm | AO à intervalle long >=3 cm |
| Approche chirurgicale | Anastomose Directe | Procédé Foker33 Thoracotomie par étapes avec croissance œsophagienne induite par tension avant anastomose |
| Troubles associés | Plus couramment associé à VACTERL35 | * Susceptible d’être une malformation isolée 97, 103 * Associé au syndrome de Down9 |
| Charge allostatique périopératoire | * Impact des anomalies coexistantes * Impact de la prématurité – le cas échéant | • Impact des anomalies chromosomiques – le cas échéant En lien avec Foker • Ventilation mécanique postopératoire prolongée • Nutrition parentérale totale prolongée • Gestion du sevrage de la sédation • Immobilité prolongée avec risque de plagiocéphalie43, 70 |
| Gestion de la douleur/sédation | <5 jours | ≥ 5 jours (associé à une dépendance physique aux médicaments sédatifs50, 92 |
| Complications postopératoires | Revues99, 104 | Risque accru de complications14, 95 CHIRURGICALES • Fuite anastomotique • Compromission respiratoire • Septicémie RÉCUPÉRATION • Sténoses et dysphagie • Reflux gastro-œsophagien • Difficultés alimentaires (c.-à-d. aversion alimentaire) |
Le tableau présente les principales différences et similitudes entre l’atrésie de l’œsophage (AO) à intervalle court et à intervalle long en ce qui concerne la présentation, l’approche chirurgicale et les complications périopératoires. Il convient de noter que les nouveau-nés recevant des soins intensifs sont susceptibles de développer une plagiocéphalie positionnelle43, une affection liée à des retards de développement70. Acronyme : VACTERL, Vertebral, Anorectal, Cardiac, Tracheo-Esophageal fistula, Renal, Limb abnormalities.
*Remarque : l’écart en cm représente un concept historique. Le type chirurgical est déterminé à la fois par la distance et par la capacité (ou l’incapacité) à effectuer une réparation primaire en une seule intervention chirurgicale.
3. Étiologie et diagnostic de l’atrésie de l’œsophage
3.1. Quelle est la cause de l’atrésie de l’œsophage ?
La cause de l’AO n’est pas encore entièrement comprise ; cependant, les données disponibles indiquent une origine multifactorielle impliquant des facteurs génétiques, développementaux et environnementaux.
Si l’AO se présente comme une anomalie isolée dans 38 %103 à 53,5 % des cas97, elle coexiste souvent avec d’autres malformations congénitales, fréquemment dans le cadre d’un syndrome reconnu. Par exemple, 10 % des patients atteints d’AE97 présentent une association VACTERL, qui comprend des anomalies Vertébrales, Anorectales, Cardiaques, une fistule Trachéo-œsophagienne et/ou une Eatrésie œsophagienne, des anomalies Rénales et des anomalies Limbiques. De plus, 15 à 20 % des cas surviennent dans le contexte du syndrome de CHARGE60, qui comprend un colobome, des hépatismes cardiaques, une atresie des choanes (également appelée atrésie choanale), un Retard de croissance, des anomalies génitales et des anomalies des oreilles. Les anomalies chromosomiques telles que la trisomie 18, la trisomie 219 ou les délétions 22q1122 augmentent également le risque d’EA. Par conséquent, les nourrissons atteints d’EA subissent systématiquement une évaluation génétique.
3.1.1. Mécanismes embryologiques
Au cours du développement fœtal, la trachée et l’œsophage proviennent d’un tube antérieur commun. L’hypothèse principale, connue sous le nom de « modèle de compartimentation », postule que la séparation de l’intestin antérieur se produit par plissement et remodelage des tissus29. La signalisation Sonic Hedgehog (Shh) joue un rôle crucial dans ce processus, car son absence entraîne une AE dans les modèles animaux61. De plus, des facteurs de transcription tels que Sox1, impliqué dans le développement de l’œsophage, et Nkx2.1, associé au développement de la trachée, sont également impliqués.
Les modèles knock-out de l’un ou l’autre de ces gènes conduisent à une AO, avec ou sans FTE46, 74.
3.1.2. Facteurs génétiques
Le syndrome de Feingold, causé par des mutations du gène NMYC, est la forme familiale d’AO la mieux caractérisée, touchant environ 40 % des personnes atteintes de cette maladie12.
D’autres troubles génétiques associés à l’AO comprennent les mutations liées au BRCA dans l’anémie de Fanconi23, les mutations CDH1108 et la surexpression de CHD7 dans le syndrome de CHARGE49. Ce dernier est un trouble autosomique dominant rare caractérisé par une grande variabilité clinique, où l’AO est considérée comme une caractéristique mineure102. De plus, des mutations dans FANCB ont été associées à l’AO, bien que leur rôle précis fasse encore l’objet d’études.
3.1.3. Interactions gènes-environnement
Les expositions environnementales peuvent exacerber la susceptibilité génétique. Par exemple, le gène Glutathione S-Transferase mu 1 (GSTM1) code pour des enzymes qui détoxifient les polluants environnementaux dans le liquide amniotique87. Une étude a montré que les fœtus dépourvus d’allèles fonctionnels GSTM1 ont un risque plus que double d’AO31. Cette découverte soutient un modèle multifactoriel impliquant à la fois des influences environnementales et génétiques.
3.1.4. Influences environnementales
Plusieurs expositions maternelles ont été associées à un risque accru d’AO, notamment le tabagisme, la consommation d’alcool110 et le contact avec des pesticides30. Une méta-analyse a également identifié la grippe maternelle comme un facteur contributif possible63. En outre, l’utilisation de certains médicaments au cours du premier trimestre, tels que le méthimazole16, l’œstrogène exogène ou la progestérone81, peut perturber le développement embryonnaire et contribuer à la formation d’une AO.
3.2. Quel est le risque d’atrésie de l’œsophage lors de grossesses futures ?
Le risque d’AO lors de grossesses futures dépend du fait que l’anomalie se présente de manière isolée ou dans le cadre d’un syndrome génétique. L’AO syndromique comporte un risque de récidive plus élevé, tandis que les cas isolés présentent généralement un taux de récidive familiale très faible.
L’AO a été décrite dans plus de 70 syndromes génétiques (tableau 1 dans11), et la récurrence dépend du mode de transmission héréditaire et de la probabilité que l’AE se manifeste dans le cadre du syndrome. Par exemple, le syndrome de CHARGE suit un mode de transmission autosomique dominant, un parent atteint ayant 50 % de chances de transmettre la maladie, bien que seulement 10 à 17 % des personnes atteintes développent une AO49, 59. L’anémie de Fanconi, en revanche, est autosomique récessive, avec un risque de récurrence de 25 % si les deux parents sont porteurs, mais l’AO ne survient que dans environ 14 % des cas23. Pour les familles touchées par ces maladies, un conseil génétique est fortement recommandé afin de clarifier les risques de récurrence et d’orienter les décisions en matière de reproduction.
Dans les cas d’AO isolée, le risque de récurrence est beaucoup plus faible et n’est pas défini avec précision. On pense qu’elle résulte de mutations sporadiques, éventuellement influencées par des expositions environnementales, plutôt que de traits génétiques héréditaires11. La récurrence chez les frères et sœurs est estimée à seulement 1 à 3 %15, certaines études ne rapportant aucun cas chez les frères et sœurs, mais des cas occasionnels chez des parents éloignés. Les taux plus élevés chez les jumeaux monozygotes par rapport aux jumeaux dizygotes90 suggèrent en outre une contribution génétique qui reste mal comprise.
3.3. Comment diagnostique-t-on l’atrésie de l’œsophage ?
Dans certains cas, l’AO peut être suspectée pour la première fois pendant la grossesse si une échographie fœtale de routine révèle un polyhydramnios (excès de liquide amniotique) ou une absence de bulle stomacale fœtale, deux éléments qui peuvent indiquer une altération de la déglutition fœtale. Après la naissance, la suspicion clinique est renforcée lorsque le nouveau-né présente la « triade classique » : (1) salive mousseuse ou « bulles », due à une déglutition inefficace ; (2) vomissements, car les aliments et les liquides ne peuvent pas passer par l’œsophage atrésique ; et (3) toux ou étouffement pendant l’alimentation, souvent accompagnés d’une cyanose, c’est-à-dire une coloration bleuâtre des lèvres et de la peau. L’une des procédures de diagnostic précoce les plus fiables consiste à tenter d’introduire une sonde nasogastrique (SNG). L’impossibilité de faire passer la sonde au-delà de 10 à 12 cm de la gencive suggère un œsophage en cul-de-sac. Une radiographie thoracique peut alors confirmer le diagnostic en montrant la SNG enroulée dans la poche œsophagienne supérieure et une bulle gastrique petite ou absente.
La présence de gaz gastrique suggère une FTE concomitante, car la fistule permet le passage de l’air dans l’estomac.
Le diagnostic définitif repose sur une combinaison d’imagerie prénatale, de résultats cliniques postnataux et de confirmation radiographique, d’autant plus que le diagnostic peut être retardé dans les cas atypiques ou plus légers, où les symptômes ressemblent à ceux du reflux gastro-œsophagien (RGO) ou de la dysphagie oropharyngée.
3.3.1. Diagnostic prénatal
Au cours du deuxième trimestre, une échographie fœtale de routine peut révéler des caractéristiques suggérant une AO, telles qu’une bulle stomacale petite ou absente et un polyhydramnios. Au troisième trimestre, un signe échographique plus spécifique, connu sous le nom de « signe de la poche », peut être observé lorsque le fœtus tente d’avaler51. La détection peut être compliquée par la présence d’une FTE83, 85, auquel cas l’imagerie par résonance magnétique (IRM) fœtale peut être utilisée. De plus, l’analyse enzymatique du liquide amniotique a démontré une grande précision diagnostique pour l’AO et n’est pas affectée par la présence d’une FTE18, 105. Malgré ces avantages, l’IRM fœtale et les tests enzymatiques sont utilisés de manière sélective. L’IRM fœtale est moins répandue et peut ne pas être réalisable dans tous les contextes cliniques, tandis que l’analyse enzymatique nécessite une amniocentèse, qui comporte un risque faible mais mesurable de complications.
3.3.2. Diagnostic postnatal
La détection prénatale restant limitée, la plupart des cas d’AO sont diagnostiqués après la naissance. Une fois la triade de symptômes reconnue, une sonde nasogastrique et une radiographie thoracique sont utilisées comme décrit ci-dessus. Après confirmation de l’AO, une évaluation complète est réalisée afin d’identifier les anomalies associées, qui surviennent dans plus de 50 % des cas. Le dépistage postnatal standard comprend une évaluation cardiaque à l’aide d’une électrocardiographie et d’une échocardiographie afin d’identifier les malformations cardiaques congénitales. Une imagerie spinale et thoracique est réalisée afin de détecter les anomalies vertébrales ou une moelle attachée, et une échographie abdominale et pelvienne est utilisée afin d’évaluer les malformations rénales et anorectales94. Notre récente analyse institutionnelle confirme que tous les nourrissons atteints d’AO subissent systématiquement une imagerie de la colonne vertébrale afin de dépister une atrésie anale et un syndrome de moelle attachée72.
3.3.3. Orientations futures en matière de diagnostic
Malgré les progrès réalisés en matière d’imagerie et de surveillance prénatale, l’AO reste difficile à diagnostiquer in utero, en particulier dans les cas bénins ou lorsqu’elle coexiste avec d’autres affections telles que le RGO. Les pratiques diagnostiques varient également d’un établissement à l’autre. Le registre européen des atrésies œsophagiennes (EUPSA-EAR) de l’Association européenne des chirurgiens pédiatriques (European Paediatric Surgeons’ Association) a mis en évidence des variations importantes entre les établissements et au sein même des établissements en matière de protocoles et de pratiques diagnostiques78. La mise en place de parcours diagnostiques prénataux standardisés pourrait améliorer la détection précoce, permettre une meilleure coordination des soins et faciliter l’orientation rapide vers des centres chirurgicaux spécialisés3. Un diagnostic précoce améliore les résultats chirurgicaux et permet aux familles de bénéficier de conseils anticipés, d’un soutien psychologique et d’une planification complète des soins avant l’accouchement.
3.4. Quelles sont les implications de l’association VACTERL ?
L’association VACTERL est définie comme un ensemble non aléatoire d’anomalies comprenant des Véhicules vertébraux, une Atresie anale, des Coronaires, une Trachéo-Esophagienne, des Rénales et/ou des Limbaires. Le diagnostic nécessite la présence d’au moins trois de ces caractéristiques. La présentation de l’association VACTERL est très variable, ce qui nécessite des stratégies de prise en charge individualisées. Les progrès des techniques chirurgicales et des soins multidisciplinaires ont considérablement amélioré les taux de survie, et la plupart des enfants atteignent des résultats de développement normaux54.
La prise en charge de l’association VACTERL est difficile en raison de son hétérogénéité et de l’absence de critères diagnostiques standardisés93. Les nourrissons subissent des évaluations complètes afin d’identifier toutes les malformations, en se concentrant dans un premier temps sur les anomalies potentiellement mortelles telles que les malformations cardiaques critiques ou la FTE.
Une fois l’état stabilisé, l’accent est mis sur les anomalies ayant des implications à long terme, telles que les malformations vertébrales. Bien que la présence des caractéristiques VACTERL ne permette pas à elle seule de prédire entièrement les résultats, elles sont associées à une mortalité accrue dans les cas d’AO/TEF et à des difficultés à long terme, notamment des retards de développement neurologique54,86,100. Un suivi multidisciplinaire continu est fortement recommandé afin d’optimiser les résultats à long terme, en particulier dans les cas d’AO à court intervalle, où les caractéristiques VACTERL sont plus fréquentes (voir tableau 1).
3.5. Le risque de prématurité est-il plus élevé chez les nourrissons atteints d’atrésie de l’œsophage ?
La prématurité, définie comme un accouchement avant 37 semaines de grossesse, est fréquente chez les nourrissons atteints d’anomalies congénitales, y compris l’AO.
Des analyses basées sur la population ont montré l’une des associations les plus fortes entre l’AO et la prématurité75, avec des taux de prématurité pouvant atteindre 37 % dans de grandes cohortes multicentriques98. Les revues institutionnelles font état de taux encore plus élevés, reflétant probablement un biais en faveur des centres tertiaires28. Les mécanismes sous-jacents à cette association sont multifactoriels. Les facteurs maternels tels que le tabagisme, la malnutrition, le stress psychosocial et les grossesses multiples sont des risques bien établis37, 89. Dans le cas de l’AO le polyhydramnios est particulièrement pertinent, car la distension utérine excessive et l’activation inflammatoire ont été impliquées dans le déclenchement du travail prématuré1, 67.
4. Réparation chirurgicale et soins intensifs
4.1. Comment planifier la réparation d’une atrésie de l’œsophage ?
Les soins optimaux pour les nourrissons atteints d’AO nécessitent une équipe multidisciplinaire dans un centre spécialisé. Les familles sont encouragées à rechercher des centres d’excellence disposant d’une expertise complète dans la prise en charge de l’AO.
Le programme de traitement de l’œsophage et des voies respiratoires
du Boston Children’s Hospital, fondé en partie par le Dr R.W. Jennings, a été le premier centre national dédié à l’AO et est aujourd’hui un leader mondial dans le domaine de la reconstruction œsophagienne complexe. Des programmes similaires ont depuis vu le jour aux États-Unis (à savoir le Children’s Hospital of Philadelphia et le Johns Hopkins All Children’s Hospital) et à l’étranger. Les familles internationales peuvent consulter The EAT Federation (Esophageal Atresia Global Support Groups) pour obtenir des conseils sur l’identification d’équipes expérimentées.27
Il est important de noter que les disparités dans l’accès aux soins spécialisés persistent tant au niveau national qu’international. Il sera essentiel de remédier à ces inégalités par le biais de collaborations institutionnelles et internationales, d’initiatives de renforcement des capacités (par exemple, formation de la main-d’œuvre, révision des politiques, amélioration continue de la qualité) et d’investissements soutenus afin d’optimiser les résultats pour tous les enfants atteints d’AO. Si la plupart des données disponibles proviennent de milieux disposant de ressources importantes, les disparités en matière de diagnostic et de suivi sont particulièrement prononcées dans les milieux disposant de ressources limitées. Les études futures devront tenir compte de ces inégalités mondiales afin de garantir que les directives de surveillance soient fondées sur des données probantes, équitables et largement applicables. En donnant la priorité à l’équité, les organismes de santé peuvent créer des réseaux plus solides et favoriser des environnements de soutien aux patients plus sûrs.
4.2. Comment l’atrésie de l’œsophage est-elle réparée ?
Tous les nourrissons atteints d’AO doivent subir une intervention chirurgicale pour rétablir la continuité de l’œsophage. L’approche chirurgicale dépend du type d’AO (Fig. 1) et de la longueur de la lacune œsophagienne. Le tableau 1 résume les principales différences entre la prise en charge de l’AO à intervalle court et celle de l’AO à intervalle long.
L’AO à intervalle court est généralement traitée par anastomose primaire, qui relie directement les segments œsophagiens peu après la naissance. L’AO à intervalle long nécessite généralement des interventions par étapes. Le procédé Foker, mis au point par le Dr John Foker33, favorise la croissance œsophagienne en appliquant des sutures de traction pendant 1 à 3 semaines avant de joindre les segments, ce qui implique souvent plusieurs thoracotomies ou procédures thoracoscopiques6.
Une revue complète de son évolution et de ses résultats est disponible ailleurs47. Les soins postopératoires comprennent la surveillance de la cicatrisation, l’évaluation de la fonction œsophagienne et la détection des complications telles que les sténoses ou le RGO. De nombreux patients atteints d’AO à intervalle long nécessitent des interventions supplémentaires au cours de la première année44. Par la suite, l’alimentation par voie orale n’est introduite qu’après confirmation de l’intégrité et du bon fonctionnement de l’œsophage.
4.3. Quel est l’impact des complications précoces et tardives après la réparation d’une atrésie de l’œsophage sur les visites de suivi ?
Bien que la réparation chirurgicale rétablisse la continuité œsophagienne, les nourrissons atteints d’AO restent exposés à des complications précoces et tardives, ce qui nécessite un suivi étroit afin d’optimiser les résultats. La fréquence et la durée de ces visites dépendent du type de réparation, de la présence de comorbidités et de l’évolution postopératoire. Les nourrissons atteints d’AO à court intervalle ont généralement des rendez-vous de suivi tous les 4 à 6 mois pendant les deux premières années. En revanche, ceux atteints d’AO à long intervalle nécessitent souvent des évaluations plus fréquentes en raison de la plus grande complexité chirurgicale et des taux de complications plus élevés. Après l’âge de deux ans, des visites annuelles sont généralement recommandées jusqu’à l’âge de cinq ans, suivies d’évaluations périodiques pendant l’adolescence afin d’évaluer la fonction œsophagienne et le développement neurologique.
Les complications précoces, survenant dans les 18 premiers mois suivant la réparation chirurgicale, sont souvent graves. Des fuites anastomotiques surviennent chez environ 15 % de tous les patients atteints d’AO19 et dans 18 % à 35 % des cas d’AO à long intervalle32. Ces fuites peuvent se manifester par des difficultés d’alimentation, de la fièvre, une augmentation du drainage ou une détresse respiratoire. Si les fuites mineures peuvent être résolues par un traitement conservateur112, les fuites importantes nécessitent souvent une nouvelle intervention chirurgicale34 ou un traitement endoscopique par aspiration66. La sténose anastomotique est la complication la plus courante, touchant 30 à 50 % des nourrissons5, généralement au cours de la première année. Elle se manifeste généralement par une dysphagie et une détresse respiratoire34, et le traitement consiste généralement en une série de dilatations œsophagiennes ou éventuellement en une révision chirurgicale52.
Les complications tardives de la réparation de l’anomalie de l’œsophage peuvent persister pendant l’enfance et à l’âge adulte, nécessitant un suivi à long terme. Le reflux gastro-œsophagien touche 40 à 79 % des patients et peut évoluer vers un RGO21, avec des symptômes tels que régurgitations, brûlures d’estomac, toux et faible prise de poids34. La prise en charge comprend des médicaments ou une intervention chirurgicale, avec une surveillance régulière recommandée indépendamment de la présence de symptômes57. Les difficultés à avaler touchent plus de 50 % des adolescents et des adultes, principalement en raison d’une dysmotilité œsophagienne et d’un péristaltisme inefficace34. Les symptômes peuvent aller de l’étouffement et de l’impaction alimentaire à une gêne thoracique. La prise en charge peut inclure des modifications alimentaires, une thérapie de la déglutition, une stimulation neuromusculaire ou la mise en place d’une sonde gastrique (sonde G).
Les problèmes respiratoires chroniques, notamment la respiration sifflante, la toux et les infections récurrentes, sont fréquents, en particulier au cours des trois premières années, et peuvent persister à l’âge adulte. Les facteurs contributifs comprennent le RGO, la dysmotilité, la trachéobronchomalacie et les fentes laryngées, qui augmentent tous le risque d’aspiration et de morbidité à long terme56. Des anomalies structurelles telles que la trachéobronchomalacie sont présentes chez environ 87 % des patients atteints d’AO, tandis que les fentes laryngées surviennent chez environ 20 % d’entre eux, contribuant toutes deux de manière significative à des complications récurrentes53, 62. La trachéomalacie touche environ 86 % des patients, mais seuls 10 % d’entre eux nécessitent une intervention2 ; les cas bénins s’améliorent souvent avec le temps et sont généralement identifiés lors d’une bronchoscopie de suivi.
Le dysfonctionnement des cordes vocales, y compris la parésie ou la paralysie, touche 3 % à 17 % des patients atteints d’AO57, 79, ce qui augmente le risque d’aspiration et d’infection chronique8, 42, en particulier dans les cas d’AO à long intervalle en raison de techniques chirurgicales complexes et d’une intubation postopératoire prolongée79. En outre, un risque plus élevé d’asthme et d’allergies a été signalé dans cette population91.
Conscients des implications chroniques et à vie de l’AO, nous soulignons la nécessité d’une transition structurée vers des soins adaptés aux adultes. Ce processus nécessite la coordination des services de gastro-entérologie, de pneumologie et de psychologie afin d’assurer la continuité de la surveillance et de la prise en charge tout au long de la vie (voir également la section 6.3 ci-dessous). Les résultats neurodéveloppementaux et la qualité de vie à long terme après la réparation de l’AO sont abordés en détail dans une autre revue.
5. Alimentation
5.1. Quels sont les défis nutritionnels et de croissance après la réparation d’une atrésie de l’œsophage ?
L’AO présente des défis nutritionnels uniques, mais des études indiquent que les nourrissons atteints peuvent atteindre des trajectoires de croissance comparables à celles de leurs pairs en bonne santé. Des équipes de soins multidisciplinaires, comprenant des chirurgiens, des pédiatres et des nutritionnistes, élaborent des plans nutritionnels individualisés afin d’optimiser la croissance. L’alimentation entérale par sonde gastrique est souvent utilisée pour contourner l’œsophage et, dans certains cas, une nutrition parentérale totale (NPT) peut être administrée par voie intraveineuse jusqu’à ce que la réparation définitive soit terminée.
Les nourrissons présentant des anomalies cardiaques congénitales ont un risque deux fois plus élevé d’être petits pour leur âge gestationnel36 et présentent souvent une prise de poids postnatale plus lente13. De même, les anomalies gastro-intestinales, y compris l’AO, augmentent le risque de faible poids à la naissance (<10e percentile)82. Lorsque la réparation chirurgicale de l’EA est retardée en raison d’une prématurité, d’une instabilité respiratoire ou de comorbidités, la prise en charge nutritionnelle est guidée par des diététiciens pédiatriques. Le soutien nutritionnel préopératoire par nutrition parentérale totale (NPT) ou entérale assure la croissance39, 40, et la mise en place d’une sonde gastrique peut être nécessaire en cas d’AO à long intervalle, en fonction du contexte clinique4, 84. Des courbes de croissance de référence sont disponibles pour guider les parents (tableau 1 dans111).
Malgré ces difficultés, la norme actuelle en matière de prise en charge nutritionnelle et alimentaire favorise généralement une croissance somatique appropriée. Plus précisément, les nourrissons subissant une réparation primaire pour une AO à écart court atteignent généralement un poids adapté à leur âge à l’âge de trois ans, malgré des déficits initiaux après la sortie de l’hôpital liés à une alimentation orale limitée, à une demande métabolique accrue et à un RGO postopératoire39. Les nourrissons atteints d’une AO à écart long prennent également un poids adéquat au fil du temps. Cependant, leur poids absolu peut rester inférieur aux normes de la population, et les patients nécessitent souvent une hospitalisation prolongée et un soutien nutritionnel prolongé, notamment une nutrition parentérale totale (NPT) et une alimentation par sonde gastrique40. Une récente étude pilote a rapporté que les nourrissons atteints d’AO à intervalle long ayant suivi le processus de Foker atteignaient des étapes de poids comparables à celles des nourrissons nés à terme et en bonne santé à l’âge d’un an7, soulignant l’importance d’un soutien nutritionnel proactif et individualisé pendant la petite enfance.
5.2. Comment la transition vers l’alimentation orale est-elle gérée après la réparation de l’atrésie de l’œsophage ?
Après la réparation de l’AO, l’objectif principal est une transition progressive vers l’alimentation orale, adaptée à l’état clinique de chaque nourrisson. Le soutien nutritionnel peut inclure une nutrition parentérale totale, une alimentation entérale ou les deux, selon les protocoles de l’établissement. Certains nourrissons commencent directement l’alimentation orale, tandis que d’autres commencent par une alimentation par sonde en petites quantités, qui est progressivement augmentée en fonction de la tolérance. Une fois que l’alimentation entérale est tolérée, des essais oraux avec du lait maternisé ou du lait maternel exprimé sont introduits et progressivement augmentés jusqu’à ce que l’alimentation orale complète soit atteinte, ce qui permet de retirer la sonde nasogastrique ou la sonde gastrique.
Après la réparation, les nourrissons reçoivent généralement une combinaison de nutrition parentérale totale et d’alimentation entérale, en évitant les sondes transanastomotiques en raison du risque de formation de sténoses58.
Les patients atteints d’AO à long intervalle commencent souvent par une nutrition parentérale totale jusqu’à ce que la longueur de l’œsophage soit suffisante pour une réparation sûre. Un œsophagogramme est généralement réalisé entre le 5e et le 7e jour postopératoire afin d’évaluer l’intégrité de l’anastomose et de déterminer si le patient est prêt pour une alimentation entérale40. La préparation à l’alimentation est souvent évaluée par un orthophoniste, qui aide à déterminer le moment opportun pour les essais oraux40.
Le sevrage de la sonde est plus efficace dans un cadre multidisciplinaire qui met l’accent sur la faim comme facteur de motivation26. Les approches comprennent la réduction progressive de l’alimentation par sonde tout en introduisant des aliments par voie orale, avec un soutien continu pour assurer une progression sûre41. Les programmes invasifs en milieu hospitalier, tels que le modèle Graz24, 69, accélèrent le sevrage de la sonde en 2 à 3 semaines, en associant une réduction supervisée de l’alimentation à une exposition orale sensorielle et ludique.
Dans les cohortes suivant cette approche, 95 % des patients atteints d’AO ont atteint une alimentation orale complète, quelle que soit la durée de l’interruption69. La plupart des patients atteints d’AO à intervalle court passent à une alimentation orale complète peu après leur sortie de l’hôpital, bien que certains aient besoin d’une alimentation supplémentaire par sonde à domicile39. En revanche, les patients atteints d’AO à intervalle long peuvent avoir besoin d’une alimentation par sonde pendant 1 à 2 ans, mais à l’âge de trois ans, presque tous parviennent à une alimentation exclusivement orale39, 40.
5.3. Quelle est la probabilité de rencontrer des difficultés alimentaires après une réparation de l’atrésie de l’œsophage ?
Les difficultés alimentaires sont courantes après une réparation de l’AO et se manifestent souvent par un refus de s’alimenter, des haut-le-cœur, des vomissements ou le fait de se détourner pendant les repas. Ces difficultés proviennent souvent d’une alimentation par sonde prolongée, d’une dysphagie ou d’un RGO. Néanmoins, la plupart des enfants franchissent avec succès les étapes clés de l’alimentation au fil du temps55.
Les taux rapportés de difficultés alimentaires après une réparation de l’AO varient considérablement en raison des différences dans les définitions et les conceptions des études106. Les estimations actuelles indiquent que 28 % à 79 % des enfants sont touchés64, 73. Ces difficultés sont plus prononcées chez les enfants présentant des anomalies associées que chez ceux présentant une AO isolée. La dysphagie touche jusqu’à 50 % des enfants atteints d’AO17, rendant l’alimentation inconfortable et décourageant l’alimentation par voie orale64.
De plus, le RGO contribue à l’inconfort, entraînant souvent une aversion pour la nourriture et des comportements d’évitement64.
Une intervention précoce ciblée peut raccourcir la durée des difficultés alimentaires, et les équipes multidisciplinaires traitent efficacement les cas complexes88. Hunt et al.44 ont décrit l’utilisation de « nourrissages simulés », dans lesquels les nourrissons tètent un sein ou un biberon tandis que la nutrition est fournie par sonde, ce qui renforce les compétences alimentaires oro-motrices et établit un lien entre la succion et la satiété. L’éducation sur les problèmes d’alimentation permet aux soignants de favoriser les progrès48, tandis que les réseaux de pairs et les groupes de parents apportent un soutien émotionnel et pratique106. Les modèles positifs, les encouragements doux et les rassurances constantes aident également les enfants à s’adapter aux difficultés d’alimentation76.
6. Sensibilisation et soutien des parents au stress
6.1. Le stress parental est-il une réaction courante au diagnostic d’AO ?
Le stress parental est une réaction courante et bien documentée au diagnostic et à la prise en charge de l’AO. Les familles sont souvent en proie à l’anxiété, à la tension émotionnelle et au fardeau psychosocial, en particulier dans la période qui suit immédiatement le diagnostic précoce et la réparation chirurgicale. Le stress est intensifié par les longs séjours en soins intensifs néonatals, l’incertitude quant au pronostic et la possibilité de complications à long terme. Le jeune âge des parents, les pressions financières et le soutien social limité augmentent encore la vulnérabilité aux problèmes de santé mentale des parents107.
Le diagnostic, généralement posé à la naissance, survient souvent de manière inattendue et provoque un sentiment de choc, de confusion et de colère.65,101 Ces réactions initiales peuvent évoluer vers des difficultés psychologiques plus persistantes, notamment l’anxiété, la dépression et les symptômes du syndrome de stress post-traumatique107, 109. Par la suite, les difficultés liées à l’alimentation et aux repas sont particulièrement pénibles et étroitement liées à l’anxiété et à la dépression des parents96, 106.
Avec le temps, le niveau de stress diminue généralement à mesure que l’enfant se stabilise et que les symptômes s’améliorent109. Compte tenu de ces risques, les équipes de soins de santé doivent identifier rapidement les familles vulnérables et leur donner accès à un soutien psychosocial. Les familles doivent également se sentir encouragées à partager ouvertement les difficultés auxquelles elles sont confrontées, en étant rassurées sur le fait que ces difficultés sont courantes et comprises par les prestataires de soins dans le cadre de l’expérience plus large qu’est l’éducation d’un enfant atteint d’AO. L’orientation vers des travailleurs sociaux, des professionnels de la santé mentale et des réseaux de parents peut contribuer à réduire le fardeau émotionnel et à améliorer le bien-être de la famille.
6.2. Quelles ressources peuvent aider les parents à gérer les difficultés après une réparation de l’atrésie de l’œsophage ?
Diverses ressources peuvent aider les parents à gérer les difficultés liées à la prise en charge d’un enfant atteint d’AO. Des soins médicaux multidisciplinaires sont essentiels, avec des pédiatres, des chirurgiens, des orthophonistes, des ergothérapeutes et des diététiciens travaillant ensemble pour répondre aux besoins complexes et évolutifs de l’enfant80.
Les programmes d’intervention précoce qui ciblent les difficultés alimentaires, les retards de développement ou les complications respiratoires améliorent encore les résultats tout en allégeant la charge des soignants80. La communication au sein de ces équipes est tout aussi importante : les interactions collaboratives et solidaires réduisent le stress parental, tandis qu’une mauvaise communication peut accroître l’anxiété et la frustration20, 68. Au-delà du système de santé, le soutien des pairs et de la communauté joue également un rôle essentiel. Les groupes de soutien aux parents favorisent les liens, réduisent l’isolement et offrent des conseils pratiques qui ne sont pas toujours disponibles dans les soins cliniques. Une récente enquête mondiale a confirmé que les réseaux de soutien dédiés à l’EA amélioraient considérablement le bien-être émotionnel des soignants et des patients20. Les plateformes en ligne élargissent ces possibilités en offrant des informations et des échanges entre pairs. Des ressources telles que l’EA/TEF Family Support Connection (EA-TEF Child and Family Support Connection)25 et The EAT Federation (Esophageal Atresia Global Support Groups)27 offrent des conseils fiables, tandis que des groupes sur les réseaux sociaux tels que Bridging the Gap of EA/TEF (sur Facebook) permettent aux familles de partager leurs expériences et de demander des conseils. Bien que ces forums soient précieux pour créer une communauté, les familles doivent vérifier toute information médicale auprès de professionnels qualifiés afin d’éviter toute désinformation.
6.3. Comment les familles peuvent-elles se préparer au mieux à un suivi à long terme dans leur établissement d’origine ?
Lors du passage d’un centre spécialisé à des soins locaux, les familles doivent établir une continuité avec des cliniciens qui connaissent bien l’AO et ses implications à long terme. Ce processus est plus efficace lorsque l’équipe tertiaire aide à identifier les prestataires et les spécialistes locaux équipés pour gérer les besoins continus.
Certaines complications, notamment les difficultés d’alimentation, le RGO et les infections respiratoires récurrentes, s’améliorent souvent avec l’âge80. D’autres, telles que la dysmotilité œsophagienne, persistent souvent et nécessitent une surveillance et une intervention continues21. C’est pourquoi un suivi à long terme est essentiel et doit être coordonné par une équipe multidisciplinaire expérimentée dans les séquelles de l’AO. Des évaluations régulières tout au long de l’enfance et de l’adolescence permettent aux cliniciens d’identifier les complications émergentes, d’orienter les interventions en temps opportun et de favoriser une croissance et un développement sains10, 45. Un calendrier de suivi structuré a été proposé afin de normaliser ce processus (Fig. 1 de45).
La continuité des soins à l’âge adulte est tout aussi essentielle. Les adolescents atteints d’AO risquent d’être perdus de vue10.
Les directives actuelles de la Société européenne de gastroentérologie, hépatologie et nutrition pédiatriques recommandent une endoscopie haute de routine à partir de 18 ans, suivie d’une surveillance tous les 5 à 10 ans ou plus tôt en cas de symptômes57. Alors que la recherche continue de clarifier les risques à long terme associés à l’AO, des plans de soins individualisés à vie seront nécessaires pour préserver la santé et la qualité de vie bien au-delà de l’enfance. En fin de compte, aider les familles à se préparer à un suivi à long terme permet non seulement de garantir la sécurité médicale, mais aussi de renforcer leur confiance, de réduire leur stress et de leur donner les moyens de naviguer dans le parcours à vie que représente l’éducation d’un enfant atteint d’atrésie de l’œsophage.
7. Conclusion
Les enfants atteints d’AO sont confrontés à des défis chroniques et multisystémiques qui vont bien au-delà de la réparation chirurgicale initiale. Si les progrès des techniques chirurgicales et des soins multidisciplinaires ont amélioré la survie, des lacunes importantes subsistent, notamment le manque de données longitudinales sur la transition vers l’âge adulte, l’intégration limitée des résultats rapportés par les parents dans les études cliniques et l’influence sous-explorée des disparités dans l’accès aux soins spécialisés.
La poursuite des recherches et la collaboration multidisciplinaire sont essentielles pour optimiser les résultats à long terme et améliorer la qualité des soins pour cette population unique. Les priorités de recherche futures devraient notamment inclure (1) la création de registres multi-institutionnels avec une curation standardisée des données, (2) l’évaluation prospective des stratégies de surveillance et (3) l’intégration de méthodes qualitatives pour saisir les perspectives des parents. Au-delà des résultats chirurgicaux, une plus grande attention doit être accordée aux trajectoires neurodéveloppementales, au soutien psychosocial et au rôle des déterminants sociaux de la santé dans la formation des résultats à long terme.
D’un point de vue clinique, les stratégies concrètes comprennent la mise en œuvre de stratégies standardisées de sevrage de la sédation et de nutrition, de protocoles de surveillance et l’intégration proactive de services de conseil parental et psychosocial, ainsi que le développement de parcours structurés de transition vers les soins pour adultes. En comblant ces lacunes et en alignant la recherche sur les priorités centrées sur la famille, le domaine peut progresser vers une prise en charge plus coordonnée et plus équitable de l’AO tout au long de la vie. Enfin, cette étude s’inscrit dans le mouvement croissant en faveur de soins centrés sur la famille en pédiatrie, qui met l’accent sur une communication personnalisée, claire et empathique. En comblant les lacunes informationnelles qui laissent les parents dépassés, nous espérons que cette ressource aidera les familles à naviguer dans la vie avec un nourrisson diagnostiqué avec une AO.